
Medical Devices

On 05.05.2020, a webinar was held in cooperation with the trade association of industrial enterprises in Baden (German: Wirtschaftsverband Industrieller Unternehmen in Baden or WIVB).
Experiences were exchanged and Cisema presented news about the current situation in China and important changes in the rules for medical devices. One focus was the introduction of the UDI system in China on 01.10.2020 (link to UDI news) and the intention of market access to the Greater Bay Area via listing in Hong Kong (link to Greater Bay Area news). Over 20 WVIB members from the German MedTech industry, followed the webinar and were able to ask detailed questions afterwards.
Link to WVIB website here.

Due to shortages of qualified medical staff and consistent with the Chinese government’s emphasis on AI technology, the NMPA has made a number of announcements providing guidance on the registration process for AI software in the healthcare sector. The most recent was in March 2020 to provide an assessment framework for product registration of Corona-related diagnostic software.
Large Chinese companies such as Tencent have also been heavily investing in AI software in the fight against Corona virus, as reported here in Chinese newspapers recently.
Software that assists with curative or diagnostic processes in China generally must be registered as a medical device with the NMPA. China has two kinds of license for AI medical devices: (1) assistive products, being those which aid healthcare professionals in their provision of care to patients; and (2) diagnostic products, being those that assist with determining the cause of an ailment.
On 05.03.2020, the Center for Medical Device Evaluation of NMPA (National Medical Products Administration) released (No.8-2020) the assessment framework for the product registration of pneumonia diagnostic software in China. The assessment applies to SaMDs (software as a medical device) adopting deep learning technology to evaluate chest CT scans, to assist triage and to diagnose clinical cases affected by the Covid-19 pandemic.
According to the NMPA, this type of software product is classified as a Class III medical device, and the software security level is B. The assessment framework advises manufacturers to focus on, but not limited to, the following requirements for the preparation of research data and supplementary materials:
Basic functions
The software should at least cover one of the following functions:
- Anomaly detection
- Quantitative analysis (For example: Lesion volume ratio, CT value distribution, etc.)
- Data comparison (Manual or automatic)
- Report output
Anomaly detection is used for the triage of suspected cases, whilst quantitative analysis and data comparison are used for the diagnosis of confirmed cases.
Data for Machine Learning
- The chest CT scan images of a minimum of 2000 Covid-19 patients
- Must be sourced from three medical institutions, and one of the medical institutions must be located in a Covid-19 heavily-affected area
- Must include the chest CT scan images of the initial and advanced stage of confirmed Covid-19 cases
Data Distribution
The data distribution of Covid-19 information based on the following factors:
- Demographics (For example: Gender and Age)
- Stages (Initial, advanced, and severe) of the chest CT scan images
- Data sourcing companies
- CT scan equipment (For example: Manufacturer and Layer thickness)
For SaMDs adopting traditional machine learning technology, the manufacturers are advised to refer to another assessment framework (No.7-2019) released by the CDME on 03.07.2019. This assessment framework applies to the product registration of SaMDs using deep learning and traditional machine learning technologies to assist medical staffs in making clinical decisions.
Both traditional machine learning and deep learning technologies are a subset of AI, however, the former normally requires human intervention for the feature extraction process, whilst the latter automatically completes the feature extraction process itself. The assessment framework addresses the basic principles of evaluating the risks, benefits, safety and effectiveness of the SaMD throughout its product lifecycle to ensure the pre-market and post-market regulatory requirements have been met.

the emergency channel relating to COVID-19 response is no longer open due to the virus situation in China coming under control.
The NMPA approved the US firm Allergan’s glaucoma drainage tube, the first imported medical device with the support of real-world data to register in China.

Advantage Austria together with Ms. Anna King from CISEMA inform about the recent regulatory and institutional developments in China.
The NMPA (National Medical Products Administration) announced the 2020 project plan and selection process for 86 medical device industry standards.
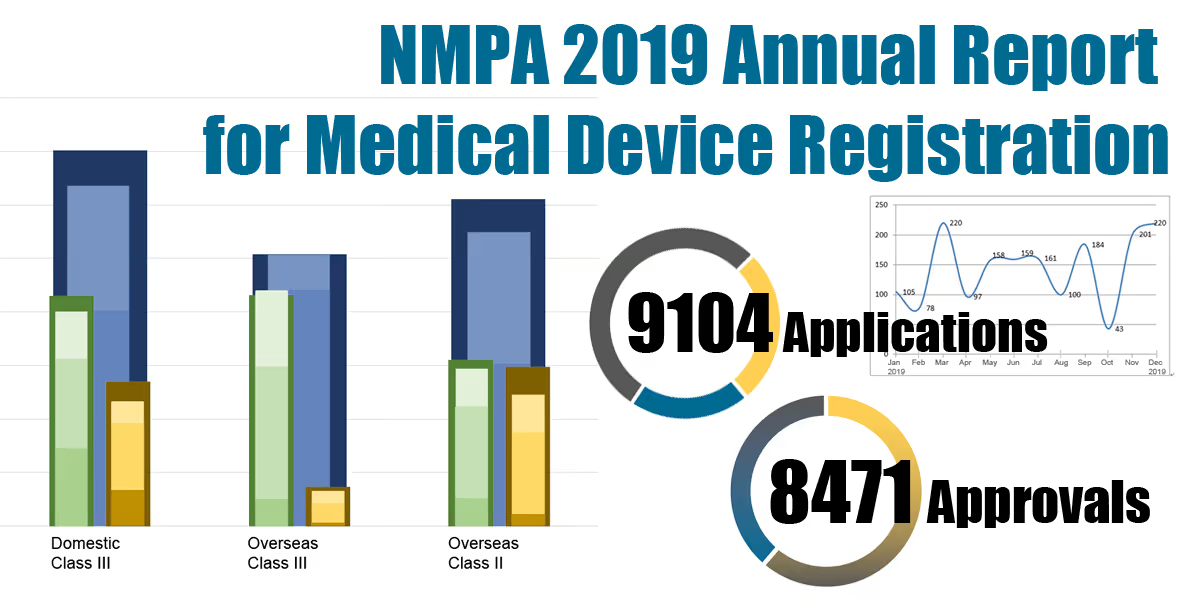
On 18.03.2020, the NMPA (National Medical Products Administration) issued the 2019 Annual Report for Medical Device Registration.In 2019, the NMPA received a total of 9,104 applications for the initial registration, registration renewals and changes in licensing items of Class III (Domestic and Overseas) and Class II (Overseas) medical devices, an increase of 37.8% from 2018. Amongst the 9,104 applications, the NMPA approved a total of 8,471 applications with an increase of 53.2% as compared to 2018. Also, the NMPA handled a total of 1,383 filing applications of imported Class I medical devices, a decrease of 20.7% as compared to 2018.
Table 1. NMPA - No. of Applications and Approvals for Medical Devices and IVDs
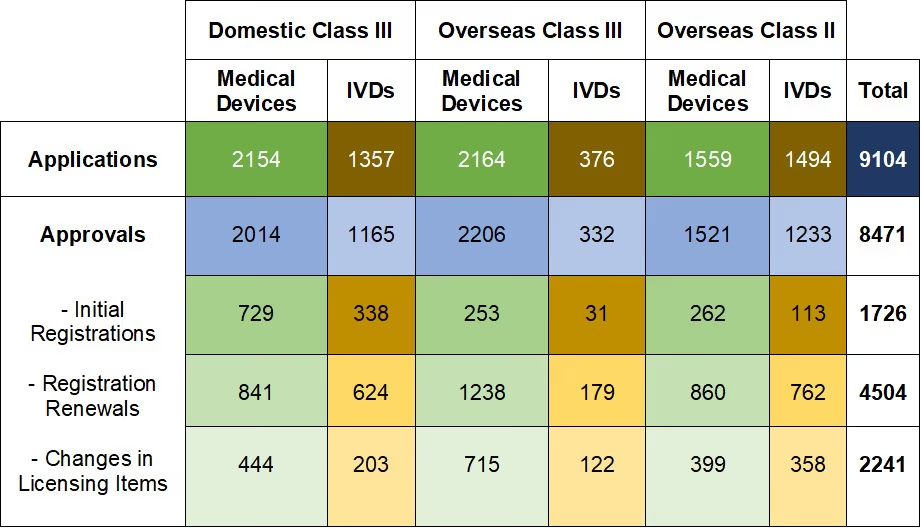
Chart 1. NMPA – No. of Applications and Approvals for Medical Devices and IVDs
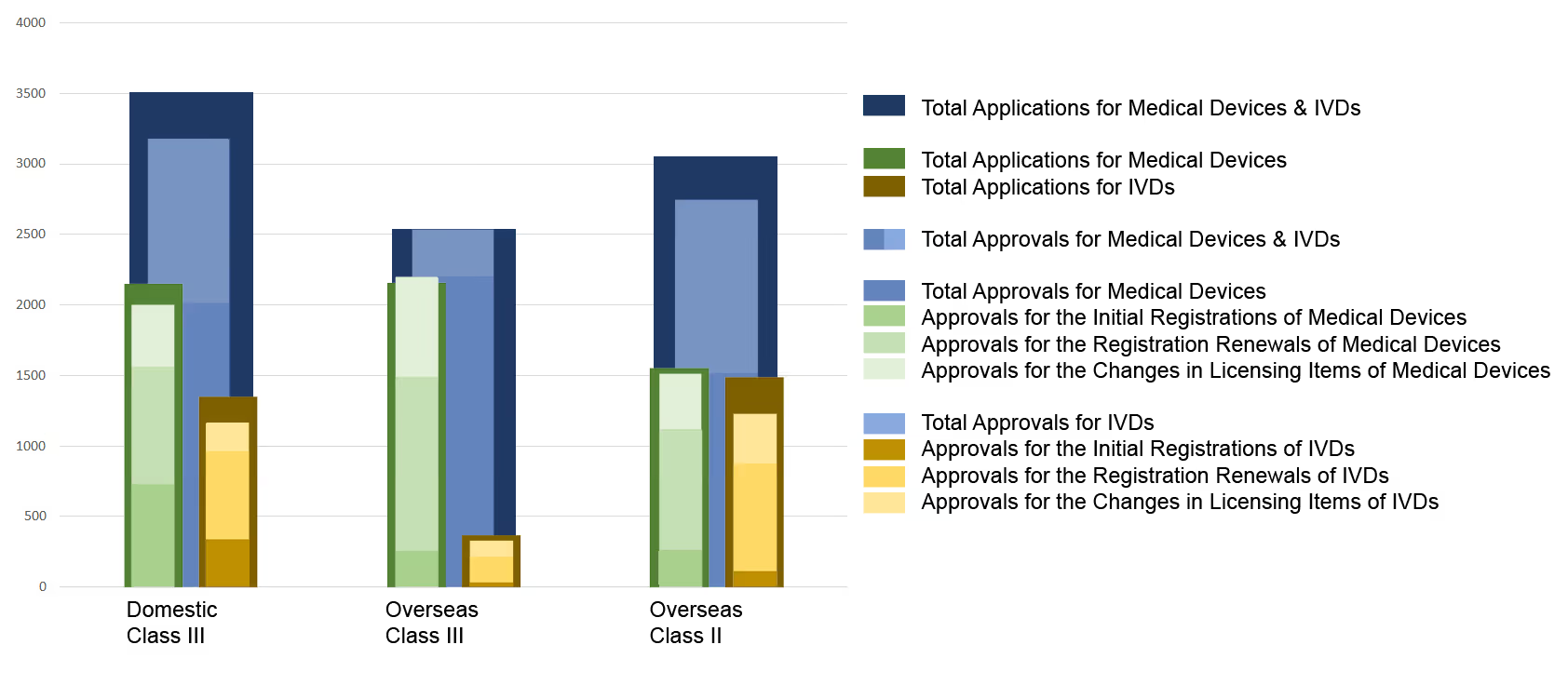
Chart 2. Percentage Distribution of Three Types of NMPA Approvals
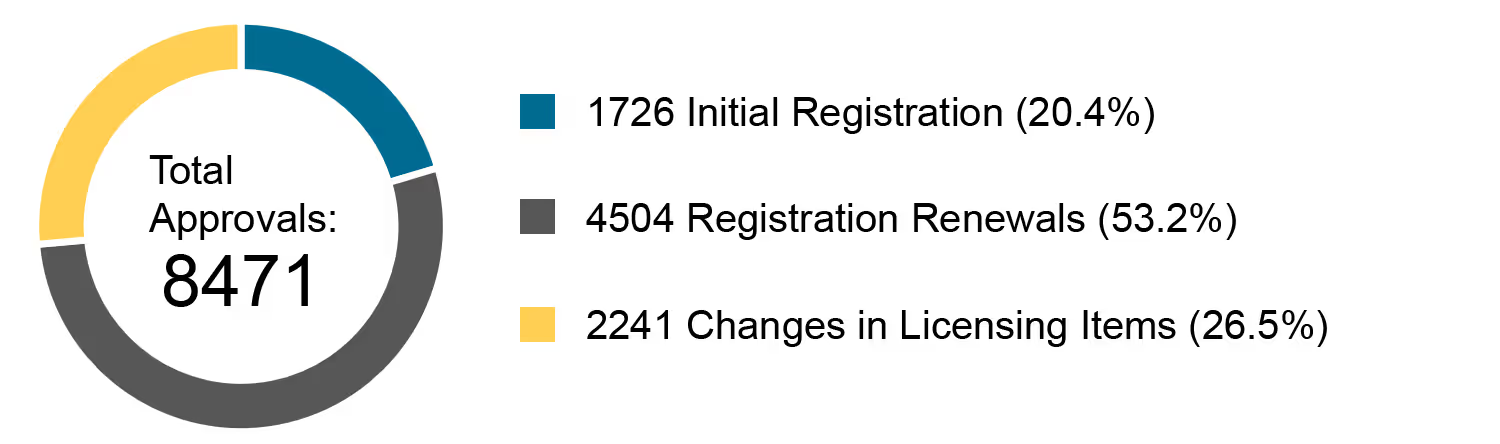
Chart 3. Percentage Distribution of Approvals for Medical Devices and IVDs

Chart 4. NMPA – No. of New Registrations Approved from January 2013 to December 2019
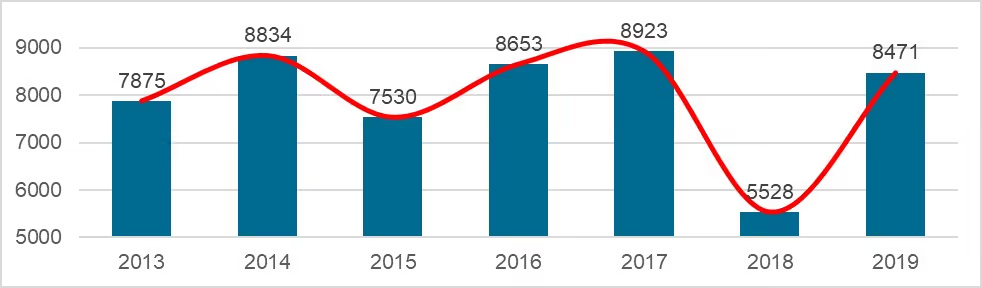
Chart 5. NMPA – No. of New Registrations Approved from January 2019 to December 2019
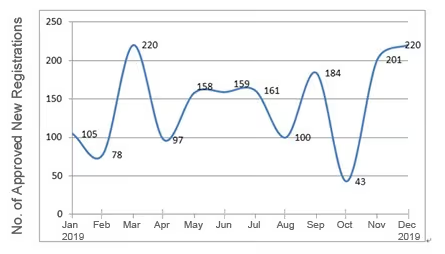
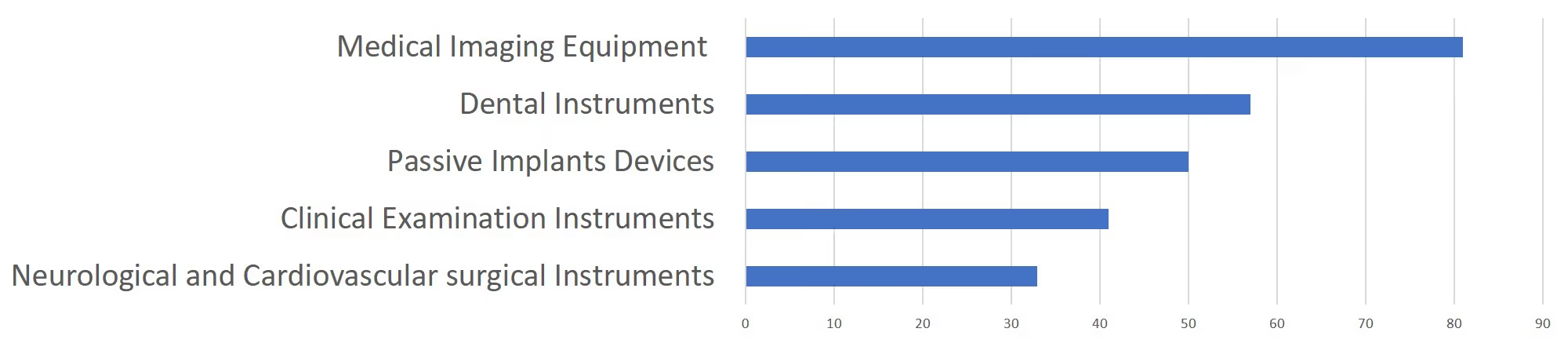
In 2019, the NMPA approved 1,726 new registrations in total.The provincial medical product administration (MPAs) authorities approved a total of 17,017 applications of China domestic Class II medical devices, an increase of 53.4% from 2018.The municipal medical product administration (MPAs) authorities handled a total of 16,754 filing applications of China domestic Class I medical devices, decreased by 2.4% as compared to 2018.China continues to focus on the supply of high-end, high-cost medical devices from abroad. The top five class II and III product groups of foreign origin to be registered in 2019 were:1. Medical Imaging Equipment (81 Registrations)2. Dental Instruments (57 Registrations)3. Passive Implants Devices (50 Registrations)4. Clinical Examination Instruments (41 Registrations)5. Neurological and Cardiovascular Surgical Instruments (33 Registrations)
Chart 6. 2019 Top 5 Product Groups - Registration of Overseas Class II and III Medical Devices
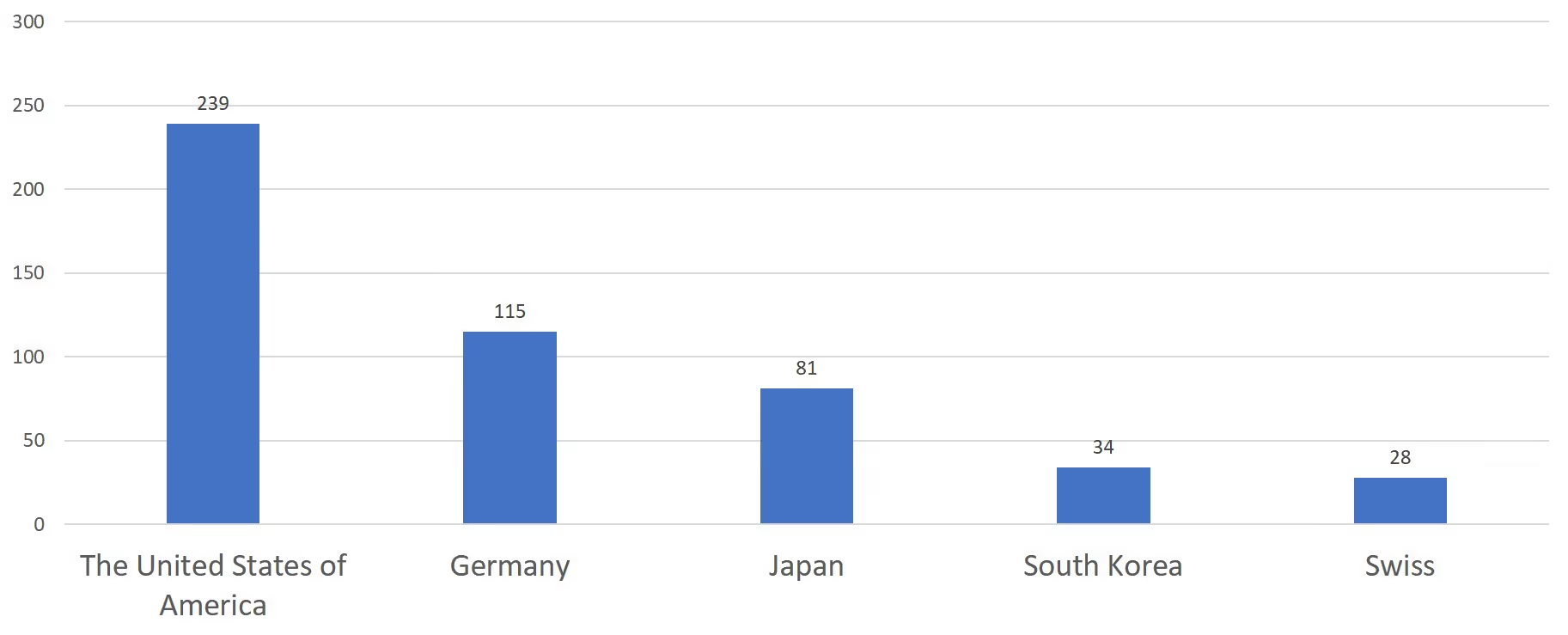
The United States of America, Germany, Japan, Korea and Switzerland have the highest number of initial registrations for overseas medical devices, taking up to 75.4% of the total number of initial registrations for overseas medical devices.
Chart 7. 2019 Top 5 Countries - Exporting Medical Devices to China
Special Review Procedures for Innovative Medical DevicesNMPA received a total of 179 applications for special review and approval of innovative medical devices, including 31 applications for priority review. A total of 19 innovative medical devices were approved:Imported1) Micra Transcatheter Leadless Pacemaker systemDomestic2) PET/CT imaging system3) Nucleic acid amplification detection analyzer4) Decellularized corneal implant5) Left atrial appendage occluder system6) Abdominal aorta stent-graft and delivery system7) Bioabsorbable coronary rapamycin-eluting stent system8) Porous tantalum bone filling material9) Patient monitor10) Left atrial appendage closure system11) Intensity-modulated radiotherapy planning system software12) Digital Mammography System13) Transcatheter aortic valve system14) Single-use intravascular imaging catheter15) Non-Invasive Blood Glucose Meter16) Implantable left ventricular assist system17) Coronary angiographic blood flow reserve fraction measurement system18) Disposable invasive pressure sensor19) Positron emission and X-ray computed tomography scanning systemFurther information concerning this topic can be obtained from:Cisema (Hong Kong) LimitedTel.: +852 3462 2483info@andrewb655.sg-host.comwww.cisema.com/en

According to the Guide, the registrant of the medical device is responsible for implementing a suitable QMS for the product realization process.
The NMPA announced new administrative measures designed to increase the post-market surveillance applicable to all registered medical devices sold in China.
The principles describe the manufacturing requirements to ensure the medical device operates safely and performs as intended。

AUSSENWIRTSCHAFT AUSTRIA and Cisema cohost the webinar to talk about the regulatory changes for both medical device and cosmetic industries.
Medical devices approved by the NMPA or the Ministry of Food and Drug Safety of Korea can apply for the MDACS listing in Hong Kong within a trial period
Get in Touch with our Certifications team
And discover how we can support you in getting your products certified for China.