
Published on
Last updated on
Understanding Digital Medical Device Clinical Trials in South Korea: The MFDS Framework

As digital healthcare technologies become increasingly data-driven and software-based, regulators are moving beyond traditional medical device frameworks to address issues such as AI validation, continuous software updates, cloud connectivity, and real-world performance monitoring.
Reflecting this shift, Korea’s Ministry of Food and Drug Safety (MFDS) established specific clinical evaluation requirements for digital medical devices under the Digital Medical Products Act. These requirements provide multiple pathways for generating clinical evidence while recognizing the unique characteristics of software-driven healthcare technologies.
The MFDS clinical evaluation framework covers:
- Digital medical device clinical trials
- Clinical performance studies
- Real-world evaluation (RWE)
- Post-market data-based studies
Importantly, the guideline also clarifies that certain data-based clinical studies and real-world evaluations may proceed with Institutional Review Board (IRB) approval alone, without requiring separate MFDS clinical trial approval under specified conditions.
For manufacturers seeking digital medical device registration in South Korea, understanding these clinical evaluation requirements is an essential step toward regulatory compliance and successful market entry.
Understanding Clinical Evaluation for Digital Medical Devices in Korea
To help manufacturers understand how these pathways apply in practice, the table below summarizes the primary clinical evaluation categories for digital medical devices that are recognized in South Korea:

Determining Applicable Clinical Trials for Digital Medical Devices
The flowchart below illustrates the decision-making process used to determine which clinical evaluation pathway may apply to a digital medical device based on its characteristics and study design.
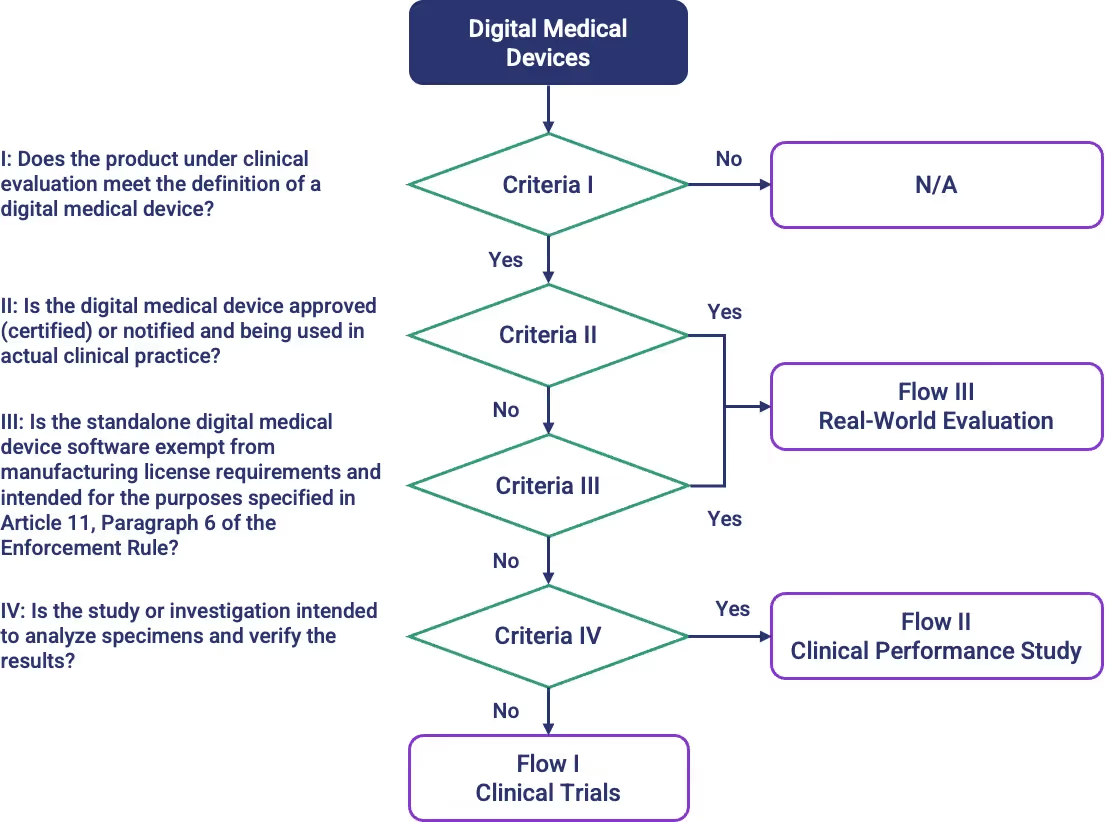
Clinical Trial Categories for Digital Medical Devices
MFDS classifies digital medical device evaluations into several categories based on product characteristics, intended use, data utilization, and risk level.
These categories include:
- Pre-market clinical trials
- Post-market clinical trials
- Clinical performance studies
- Data clinical trials
- Real-world evaluation (RWE)
Among these categories, data clinical trials are particularly important for AI medical devices and Software as a Medical Device (SaMD). These studies utilize data generated or collected through medical devices, pharmaceuticals, digital health support devices, and healthcare databases without necessarily exposing study participants to additional device-related risks.
Because these studies may not directly expose subjects to additional device-related risks, some may qualify for:
- MFDS approval exemptions
- Simplified review pathways
- IRB-only approval procedures
This approach is expected to support more flexible and efficient clinical development strategies for digital healthcare companies and AI-driven medical technologies.
Use of Real-World Evidence and Data-Based Studies
One of the most significant changes introduced by the guideline is the formal expansion of real-world evaluation (RWE) and retrospective observational research.
MFDS recognizes a broader range of post-market and data-driven clinical evaluation activities, including:
- Post-market clinical studies
- Healthcare data analysis
- Adverse event analysis
- Retrospective observational studies
- Clinical literature evaluations
Notably, some real-world evaluation activities may proceed with IRB approval alone, without separate MFDS clinical trial authorization. The approach reflects increasing regulatory acceptance of real-world data in areas such as AI model validation, software performance assessment, and post-market monitoring
The framework may be particularly relevant for manufacturers of AI diagnostic software, digital therapeutics, remote monitoring platforms, and continuously learning software systems that depend on ongoing data collection and iterative performance evaluation.
Clinical Studies Beyond Traditional Clinical Trial Institutions
MFDS recognizes that digital healthcare technologies often require cloud-based infrastructure, remote patient monitoring, decentralized data collection, and distributed healthcare environments. Therefore, under specified conditions, clinical activities may involve:
- Non-hospital institutions
- Remote participation
- Cloud platforms
- External data processing environments
- Decentralized operational models
Examples include:
- Network-based data collection
- Digital biomarker monitoring
- Decentralized AI model validation
- Remote software-based patient interaction
This approach aligns South Korea's digital medical device regulations with global trends toward decentralized clinical trials (DCTs) and digitally enabled clinical research.
Determining Eligibility for Conducting Clinical Trials Outside of Clinical Trial Institutions
The flowchart below outlines the key eligibility criteria used to determine whether a digital medical device clinical study may be conducted outside a traditional clinical trial institution.

Software Validation and Cybersecurity Expectations
Although the framework provides greater flexibility for digital clinical studies, it also raises expectations related to software quality and cybersecurity management.
The guideline emphasizes that clinical trial submissions may require:
- Software verification and validation documentation
- Cybersecurity protection measures
- Technical performance validation
- Cloud/network security controls
- Software lifecycle management documentation
For AI-based medical devices and Software as a Medical Device (SaMD), regulators may also expect evidence relating to:
- AI model training and validation
- Algorithm change management
- Software update controls
- Real-world performance monitoring
- Ongoing cybersecurity maintenance
The guideline further emphasizes the importance of IRB oversight, data governance, and continuous post-market monitoring as part of long-term compliance management for digital healthcare technologies.
Key Takeaways for Overseas Manufacturers
South Korea is continuing to build one of Asia’s most advanced regulatory frameworks for digital healthcare technologies, AI-based medical devices, and Software as a Medical Device (SaMD). The MFDS clinical evaluation requirements reflects a broader regulatory shift toward supporting real-world evidence generation, data-driven clinical evaluation models, and more flexible approaches to digital health research.
At the same time, regulatory expectations are becoming more sophisticated, particularly in areas such as software validation, cybersecurity, lifecycle management, AI transparency, and post-market monitoring.
For overseas manufacturers seeking South Korea medical device registration, successful market entry increasingly depends on:
- Early-stage regulatory strategy,
- Proper classification of digital clinical evaluation pathways,
- Local regulatory expertise,
- Continuous lifecycle compliance management.
These developments are likely to have the greatest impact on manufacturers of AI-based medical devices, digital therapeutics, remote monitoring systems, cloud-connected healthcare platforms, and other data-driven software solutions, particularly as MFDS continues to expand and refine its regulatory framework under the Digital Medical Products Act.
For manufacturers seeking guidance on Korean medical device registration, digital health compliance, and MFDS regulatory strategy, contact Cisema today.

Further Information
For guidance on Korean medical device registration and compliance requirements, manufacturers can explore Cisema’s South Korea Medical Device Registration Services.
References
- The Digital Medical Products Act (Korean)
- Regulation on Approval, Conduct, and Management of Clinical Investigation Plans for Digital Medical… (Korean)
- Good Manufacturing Practice (GMP) Requirements for Digital Medical Devices (Korean)
- Security Guideline for Protection Against Cybersecurity Threats to Digital Medical Devices (Korean)
- The Medical Devices Act (Korean)
Connect with Cisema
With more than 20 years of experience and a team of over 100 specialists, Cisema helps global companies achieve compliance across Asia Pacific with confidence and accelerate market entry.
Stay Informed with Monthly News and Analysis
Stay informed with the latest regulatory changes, expert insights, and market opportunities across APAC, delivered straight to your inbox.
Related Articles

Discover the documents required for Indonesia medical device and IVD registration, including dossier requirements, legalization, submission and common FAQs.

Understand the key components of the Malaysia Medical Device Registration Dossier for Class B, C and D device submissions.
Learn how South Korea’s Ministry of Food and Drug Safety regulates medical devices, including classification, application review and post-market duties.